| Active Ingredient | BARICITINIB |
|---|
| Drug Name | FDA Application No. | Company | Dosage Form;Route | Strength | RLD Strength | Original Approval or Tentative Approval Date |
Exclusivity Expiration (NCE) |
Exclusivity Expiration (ODE) |
Chemical Type |
Review Classification |
Marketing Status |
TE Code |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OLUMIANT | 207924 | ELI LILLY AND CO | TABLET;ORAL | 2MG | 2MG | May 31, 2018 | May 31, 2023 | _ | Type 1 - New Molecular Entity | STANDARD | Prescription | None |
| Parameters | Details |
|---|---|
| Structural Formula |
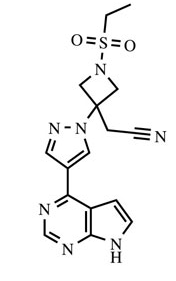 |
| Chemical Name | {1-(ethylsulfonyl)-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]azetidin-3-yl}acetonitrile |
| CAS No | 1187594-09-7 |
| Molecular Formula | C16H17N7O2S |
| Molecular Weight | 371.42 |
| Appearance | Practically white to light pink powder |
| Solubility | It is very slightly soluble in ethanol and sparingly soluble in acetone and tetrahydrofuran |
| Water Solubility | Practically insoluble in water and slighlty soluble in lower pH |
| Polymorphism | Several crystalline forms of baricitinib free base were observed during a comprehensive polymorph screen. Crystalline form I of baricitinib free base, a thermodynamically stable anhydrous form, was selected for commercial development. |
| pKa (Strongest Acidic) | 13.89 |
| pKa (Strongest Basic) | 3.91 |
| Log P | 1.08 |
| Identification | by IR |
| Degradation | Baricitinib active substance in solid state did not exhibit any detectable degradation under the stressed conditions of heat, heat and humidity, or simulated sunlight |
| Hygroscopic | Non-hygroscopic |
| Photostability study | Not sensitive to light |
| Melting Point | - |
| BCS Class | III |
| Manufacture of API | Baricitinib is synthesized using a convergent synthesis,consisting of three sequential coupling reactions of the starting materials, with requisite protection and deprotection steps to ensure proper connectivity. The final step involves a deprotection reaction to form the active substance. Designation of the staring materials is in line with the scientific advice sought from the CHMP prior to submission of the dossier. Additional data requested at the time of the scientific advice procedure has been provided and it was considered acceptable. |
| Parameters | Details |
|---|---|
| Indications and Usage | OLUMIANT® is a Janus kinase (JAK) inhibitor indicated for the treatment of adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response to one or more TNF antagonist therapies. Limitation of Use: Use of OLUMIANT in combination with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants such as azathioprine and cyclosporine is not recommended. |
| Dosage and Administration |
The recommended dose of OLUMIANT is 2 mg once daily. OLUMIANT may be used as monotherapy or in combination with methotrexate or other DMARDs. Anemia: Avoid initiation or interrupt OLUMIANT in patients with hemoglobin less than 8 g/dL. Lymphopenia: Avoid initiation or interrupt OLUMIANT in patients with an Absolute Lymphocyte Count less than 500 cells/mm3 |
| Mechanism of action |
Baricitinib is a Janus kinase (JAK) inhibitor. JAKs are intracellular enzymes which transmit signals arising from cytokine or growth factor-receptor interactions on the cellular membrane to influence cellular processes of hematopoiesis and immune cell function. Within the signaling pathway, JAKs phosphorylate and activate Signal Transducers and Activators of Transcription (STATs) which modulate intracellular activity including gene expression. Baricitinib modulates the signaling pathway at the point of JAKs, preventing the phosphorylation and activation of STATs. JAK enzymes transmit cytokine signaling through their pairing (e.g., JAK1/JAK2, JAK1/JAK3, JAK1/TYK2, JAK2/JAK2, JAK2/TYK2). In cell-free isolated enzyme assays, baricitinib had greater inhibitory potency at JAK1, JAK2 and TYK2 relative to JAK3. In human leukocytes, baricitinib inhibited cytokine induced STAT phosphorylation mediated by JAK1/JAK2, JAK1/JAK3, JAK1/TYK2, or JAK2/TYK2 with comparable potencies. However, the relevance of inhibition of specific JAK enzymes to therapeutic effectiveness is not currently known. |
| Absorption |
Following oral administration of OLUMIANT, peak plasma concentrations are reached approximately at 1 hour. A dose-proportional increase in systemic exposure was observed in the therapeutic dose range. The pharmacokinetics of baricitinib do not change over time. Steady-state concentrations are achieved in 2 to 3 days with minimal accumulation after once-daily administration. Absorption – The absolute bioavailability of baricitinib is approximately 80%. An assessment of food effects in healthy subjects showed that a high-fat meal decreased the mean AUC and Cmax of baricitinib by approximately 11% and 18%, respectively, and delayed the tmax by 0.5 hours. Administration with meals is not associated with a clinically relevant effect on exposure. In clinical studies, OLUMIANT was administered without regard to meals. |
| Food Effect | An assessment of food effects in healthy subjects showed that a high-fat meal decreased the mean AUC and Cmax of baricitinib by approximately 11% and 18%, respectively, and delayed the tmax by 0.5 hours. Administration with meals is not associated with a clinically relevant effect on exposure. In clinical studies, OLUMIANT was administered without regard to meals. |
| Distribution | After intravenous administration, the volume of distribution is 76 L, indicating distribution of baricitinib into tissues. Baricitinib is approximately 50% bound to plasma proteins and 45% bound to serum proteins. Baricitinib is a substrate of the Pgp, BCRP, OAT3 and MATE2-K transporters, which play roles in drug distribution. |
| Metabolism | Approximately 6% of the orally administered baricitinib dose is identified as metabolites (three from urine and one from feces), with CYP3A4 identified as the main metabolizing enzyme. No metabolites of baricitinib were quantifiable in plasma. |
| Elimination |
The total body clearance of baricitinib is 8.9 L/h in patients with RA. Elimination half-life in patients with rheumatoid arthritis is approximately 12 hours. Excretion – Renal elimination is the principal clearance mechanism for baricitinib through filtration and active secretion as baricitinib is identified as a substrate of OAT3, Pgp, BCRP and MATE2-K from in vitro studies. In a clinical pharmacology study, approximately 75% of the administered dose was eliminated in the urine, while about 20% of the dose was eliminated in the feces. Baricitinib was excreted predominately as unchanged drug in urine (69%) and feces (15%). |
| Peak plasma time (Tmax) | 1 hour |
| Half life | In patients with rheumatoid arthritis is approximately 12 hours |
| Bioavailability | 80% |
| Age, gender | Body weight, gender, race, ethnicity, and age did not have a clinically relevant effect on the PK (AUC and Cmax) of baricitinib (Figure 1). The mean effects of intrinsic factors on PK parameters (AUC and Cmax) were generally within the inter-subject PK variability of baricitinib. The inter-subject variabilities (% coefficients of variation) in AUC and Cmax of baricitinib are approximately 41% and 22%, respectively. |
| DMF | Status | Type | Submit Date | Holder |
|---|---|---|---|---|
| Not Available | ||||
| Parameters | Details | ||
|---|---|---|---|
| Strength | 2 MG | 4 MG | |
| Excipients used | Croscarmellose sodium, magnesium stearate, mannitol, microcrystalline cellulose | ||
| Composition of coating material | Ferric oxide, lecithin (soya), polyethylene glycol, polyvinyl alcohol, talc and titanium dioxide. | ||
| Composition of caspule shell | _ | ||
| Pharmaceutical Development |
Totale tablet weigh 206 mg. Dissolution method is sensitive to difference in drug substance particle size and change in performance due to moisture exposure during storage at ACC. Significant changes in drug substance from phosphate salt to free base and formulation capsule to tablet did not results in an change in in vivo exposure. |
||
| Manufacture of the product |
Blending steps, roller compaction, sizing, final blending, tablet compression, film-coating and packaging step |
||
| Tablet / Capsule Image | |||
| Appearance | Light pink, oblong, debossed with “Lilly” on one side and “2” on the other. | Medium pink, 8.5 mm round tablets, debossed with “Lilly” on one side and “4” on the other. | |
| Imprint code / Engraving / Debossment | Debossed with “Lilly” on one side and “2” on the other. | Debossed with “Lilly” on one side and “4” on the other. | |
| Score | No score | No score | |
| Color | Light Pink | Medium Pink | |
| Shape | OBLONG | Round | |
| Dimension | 9.0 x 7.5 mm | 8.5 mm | |
| Mfg by | - | ||
| Mfg for | - | ||
| Marketed by | Lilly USA, LLC, Indianapolis, IN 46285, USA | ||
| Distributed by | - | ||
| Application No. | Prod No | Patent No | Patent Expiration | Drug Substance Claim | Drug Product Claim | Patent Use Code | Delist Requested | Link |
|---|---|---|---|---|---|---|---|---|
| N207924 | 1 | 8158616 | June 8, 2030 | DS | DP | - | - | Download |
| N207924 | 1 | 8420629 | March 10, 2029 | - | - | U-247 | - | Download |
| USP Apparatus | Speed (RPMs) | Medium | Volume (mL) | Recommended Sampling Times (minutes) | Date Updated |
|---|---|---|---|---|---|
| II (Paddle) | 75 | pH 4.5 Acetate buffer | 900 mL | Q point 30 min | As per SBOA |
| Label | Link |
|---|---|
| FDA label | Download |
| FDA chemistry review | Download |
| FDA Pharmacology Review(s) | Download |
| FDA Clinical Pharmacology Biopharmaceutics Review(s) | Download |
| FDA BE Recommendation | |
| European Public Assessment Report | Download |
| Territory | Brand name / Generic company name | Link |
|---|---|---|
| EU | Olumiant | Download |
| UK | - | |
| US | OLUMIANT | Download |
| Date of first Authorisation in EU: 13 February 2017 |
| www.accessdata.fda.gov, www.drugbank.ca, www.ema.europa.eu, www.medicines.org.uk, dailymed.nlm.nih.gov |