| Active Ingredient | APALUTAMIDE |
|---|
| Drug Name | FDA Application No. | Company | Dosage Form;Route | Strength | RLD Strength | Original Approval or Tentative Approval Date |
Exclusivity Expiration (NCE) |
Exclusivity Expiration (ODE) |
Chemical Type |
Review Classification |
Marketing Status |
TE Code |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ERLEADA | (NDA): 210951 | JANSSEN BIOTECH | TABLET;ORAL | 60MG | 60MG | February 14, 2018 | February 14, 2023 | _ | Type 1 - New Molecular Entity | PRIORITY | Prescription | No |
| Parameters | Details |
|---|---|
| Structural Formula |
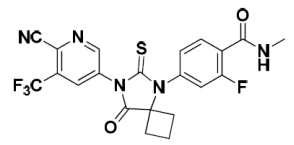 |
| Chemical Name | (4-[7-(6-Cyano-5-trifluoromethylpyridin-3-yl)-8-oxo-6-thioxo-5,7-diazaspiro[3.4]oct-5-yl]-2-fluoro-N-methylbenzamide) |
| CAS No | 956104-40-8 |
| Molecular Formula | C21H15F4N5O2S |
| Molecular Weight | 477.44 |
| Appearance | White to slightly yellow powder |
| Solubility | Practically insoluble in aqueous media over a wide range of pH values |
| Water Solubility | 0.00178 mg/mL (Predicted) |
| Polymorphism | It exhibits polymorphism, with Form B being the thermodynamically most stable form of apalutamide under the relevant crystallization and storage conditions of the active substance.The active substance synthesis process was designed to consistently deliver Form B. |
| pKa (Strongest Acidic) | 9.7 (acidic carboxamide moiety) |
| pKa (Strongest Basic) | -0.75 (Predicted) |
| Log P | 2.89 (pH 7.0) |
| Identification | IR |
| Degradation | Forced degradation studies on the active substance in solution under stress conditions of thermal acidic, thermal alkaline, thermal oxidative, neutral, dry heat, humid heat, and metal ions were performed. Results from the forced degradation studies including assay and chromatographic purity results and mass balance calculations in the form of table were presented. The active substance was found stable under neutral conditions and when exposed to metal ions. Apalutamide is prone to minor degradation under strong acidic and thermal conditions and unstable under alkaline and strong oxidative conditions. The primary degradation process of the active substance was described and the stability indicating nature of the UHPLC method for related substance and assay was shown |
| Hygroscopic | Nonhygroscopic |
| Photostability study | Photostability was investigated according to ICH Q1B. Both protected and unprotected samples were stable when exposed to light. Nevertheless, the applicant wishes to apply the storage condition “store in original package to protect from light” as a precaution measure. As there is no impact to the end user of the finished product neither to the pharmaceutical distribution chain, no objection is raised and the proposed storage condition is accepted. |
| Melting Point | 194-196 °C |
| BCS Class | BCS Class II |
| Manufacture of API | Apalutamide is manufactured through a five step synthesis reaction and purification process. The choice of the starting materials (SMs) is in line with previous CHMP Scientific Advice and the justification for all three was provided in accordance to ICH Q11. They were therefore considered acceptable and are controlled by appropriate specifications. The mutagenicity assessment showed that two impurities belong to the ICH M7 class 3. For both impurities it has been shown that they are satisfactorily controlled. Two solvents are known carcinogenic or mutagenic compounds, but based on purging factor considerations under the applicable process conditions they are not expected to occur in the final active substance at levels above their acceptable limits, and therefore do not require specific control actions. Two different synthetic routes were used during process development: synthesis method 1 and synthesis method 2. The synthesis method 1 was not a commercially viable route; therefore synthesis method 2 was developed. Three different versions of synthesis method 2 were developed. The commercial route of apalutamide active substance is clearly defined. Differences between the different versions of synthesismethods were discussed and justified. Apalutamide active substance is packaged in double, antistatic, low-density polyethylene (LDPE) bags, which are closed appropriately with a twist-tie or equivalent. The bags are placed in a closed container (plastic drum,fiber drum, or equivalent). The container closure system intended for commercial packaging of the active substance complies with the current European guideline on Plastic Immediate Packaging Materials CPMP/QWP/4359/03, including Regulation (EU) No 10/2011 on Plastic Materials and Articles Intended to Come into Contact with Food. The packaging material is controlled by acceptable in house specification. The suitability of this container closure system is demonstrated by the active substance stability data. |
| Parameters | Details |
|---|---|
| Indications and Usage | ERLEADA is an androgen receptor inhibitor indicated for the treatment of patients with non-metastatic castration-resistant prostate cancer. |
| Dosage and Administration | ERLEADA 240 mg (four 60 mg tablets) administered orally once daily. Swallow tablets whole. ERLEADA can be taken with or without food. Patients should also receive a gonadotropin-releasing hormone (GnRH) analog concurrently or should have had bilateral orchiectomy. |
| Mechanism of action |
Apalutamide is an Androgen Receptor (AR) inhibitor that binds directly to the ligand-binding domain of the AR. Apalutamide inhibits AR nuclear translocation, inhibits DNA binding, and impedes AR-mediated transcription. A major metabolite, N-desmethyl apalutamide, is a less potent inhibitor of AR, and exhibited one-third the activity of apalutamide in an in vitro transcriptional reporter assay. Apalutamide administration caused decreased tumor cell proliferation and increased apoptosis leading to decreased tumor volume in mouse xenograft models of prostate cancer. |
| Absorption |
Pharmacokinetics Apalutamide pharmacokinetic parameters are presented as the mean [standard deviation (SD)] unless otherwise specified. Apalutamide Cmax and area under the concentration curve (AUC) increased proportionally following repeated once-daily dosing of 30 to 480 mg (0.125 to 2 times the recommended dosage). Following administration of the recommended dosage, apalutamide steady-state was achieved after 4 weeks and the mean accumulation ratio was approximately 5-fold. Apalutamide Cmax was 6.0 mcg/mL (1.7) and AUC was 100 mcg·h/mL (32) at steady-state.Daily fluctuations in apalutamide plasma concentrations were low, with mean peak-to-trough ratio of 1.63. An increase in apparent clearance (CL/F) was observed with repeat dosing, likely due to induction of apalutamide’s own metabolism. The auto-induction effect likely reached its maximum at the recommended dosage because exposure of apalutamide across the dose range of 30 to 480 mg is dose-proportional. The major active metabolite N-desmethyl apalutamide Cmax was 5.9 mcg/mL (1.0) and AUC was 124 mcg·h/mL (23) at steady-state after the recommended dosage. N-desmethyl apalutamide was characterized by a flat concentration-time profile at steady-state with a mean peak-to-trough ratio of 1.27. Mean AUC metabolite/parent drug ratio for N-desmethyl apalutamide following repeat-dose administration was 1.3. Based on systemic exposure, relative potency, and pharmacokinetic properties, N-desmethyl apalutamide likely contributed to the clinical activity of apalutamide. Absorption Mean absolute oral bioavailability was approximately 100%. Median time to achieve peak plasma concentration (tmax) was 2 hours (range: 1 to 5 hours). |
| Food Effect | Administration of apalutamide to healthy subjects under fasting conditions and with a high-fat meal (approximately 500 to 600 fat calories, 250 carbohydrate calories, and 150 protein calories) resulted in no clinically relevant changes in Cmax and AUC. Median time to reach tmax was delayed approximately 2 hours with food. |
| Distribution | The mean apparent volume of distribution at steady-state of apalutamide was approximately 276 L. Apalutamide was 96% and N-desmethyl apalutamide was 95% bound to plasma proteins with no concentration dependency. |
| Metabolism |
Metabolism is the main route of elimination of apalutamide. Apalutamide is primarily metabolized by CYP2C8 and CYP3A4 to form active metabolite, N-desmethyl apalutamide. The contribution of CYP2C8 and CYP3A4 in the metabolism of apalutamide is estimated to be 58% and 13% following single dose but changes to 40% and 37%, respectively at steady-state. Apalutamide represented 45% and N-desmethyl apalutamide represented 44% of the total AUC following a single oral administration of radiolabeled apalutamide 240 mg. |
| Elimination |
Elimination The CL/F of apalutamide was 1.3 L/h after single dosing and increased to 2.0 L/h at steady-state after once-daily dosing likely due to CYP3A4 auto-induction. The mean effective half-life for apalutamide in patients was approximately 3 days at steady-state. Excretion Up to 70 days following a single oral administration of radiolabeled apalutamide, 65% of the dose was recovered in urine (1.2% of dose as unchanged apalutamide and 2.7% as N-desmethyl apalutamide) and 24% was recovered in feces (1.5% of dose as unchanged apalutamide and 2% as N-desmethyl apalutamide) |
| Peak plasma time (Tmax) | Fasting : 2 hours (range: 1 to 5 hours) With Food: delayed approximately 2 hours |
| Half life | Approximately 3 days at steady-state |
| Bioavailability | 100% |
| Age, gender |
No clinically significant differences in the pharmacokinetics of apalutamide or N-desmethyl apalutamide were observed based on age (18-94 years), race (Black, non-Japanese Asian,Japanese), mild to moderate (eGFR 30-89 mL/min/1.73m2 , estimated by the modification of diet in renal disease [MDRD] equation) renal impairment, or mild (Child-Pugh A) to moderate(Child-Pugh B) hepatic impairment. The effect of severe renal impairment or end stage renal disease (eGFR ≤29 mL/min/1.73m2 ,MDRD) or severe hepatic impairment (Child-Pugh C) on apalutamide pharmacokinetics is unknown. |
| DMF | Status | Type | Submit Date | Holder |
|---|---|---|---|---|
| Not Available | ||||
| Parameters | Details |
|---|---|
| Strength | 60 MG |
| Excipients used |
Colloidal anhydrous silica, croscarmellose sodium, hydroxypropyl methylcellulose-acetate succinate, magnesium stearate,microcrystalline cellulose, and silicified microcrystalline cellulose |
| Composition of coating material |
Iron oxide black, iron oxide yellow, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide. |
| Composition of caspule shell | NA |
| Pharmaceutical Development |
The Quality Target Product Profile (QTPP) for Erleada was defined as an immediate-release, oral, film-coated tablet, containing minimum 60 mg of apalutamide for dosing a maximum of 4 tablets a day, enabling a daily dose of 240 mg; the finished product must have a sufficiently low level of impurities and microbial burden, with a shelf life of minimum 24 months when packaged in blisters or bottles and stored at room temperature. Based on the low aqueous solubility observed across the studied pH range, and high intestinal permeability, the active substance is a class 2 compound according to the biopharmaceutical classification system (BCS) for the maximum dose of 240 mg per intake as per the SmPC. The low solubility of active substance (AS) in aqueous media was the main challenge of formulation evelopment. In order to improve the bioavailability of this BCS Class 2 substance in Erleada, development focused on increasing the active substance’s aqueous solubility and dissolution rate. Ensuring and maintaining the solid state of the intermediate is important for the bioavailability of the product.Therefore, the solid state of the intermediate has been monitored extensively at release and during stability studies of the intermediate and of the finished product throughout development. The solid state of apalutamide in the intermediate was characterised on representative batches manufactured by the development site as well as the commercial site. Different techniques, including powder X-ray diffraction (XRD), infrared spectroscopy (IR), modulated temperature differential scanning calorimetry (MTDSC), dynamic vapour sorption (DVS) and near infrared spectroscopy (NIR), were used. Data was also collected during storage at 25 °C/60% RH, 30 °C/75% RH, and 40 °C/75% RH when packaged in the proposed container closure system (low-density polyethylene bag in an aluminum laminated bag). All available data show that the intermediate is stable during storage of the intermediate and during the manufacture and storage of the finished product in the proposed container closure system. It has been demonstrated that the particle size distribution of the active substance does not impact the manufacturability, quality, or performance of the finished product and that the particle size range studied of the intermediate does not have an impact on the in vivo performance of the finished product. The development of the dissolution method was described sufficiently and was subject of Scientific Advice given by CHMP. The proposed dissolution method has been optimised for the following parameters: medium pH, type of surfactant and its concentration in the medium and the paddle rotation speed. The developed dissolution method has demonstrated to be able to provide discriminating capabilities towards certain material attributes and process parameters and storage. Considering the provided information, the CHMP agreed that the proposed dissolution method is suitable for the routine quality control of Erleada finished product. |
| Manufacture of the product |
Erleada film coated tablets manufacturing process of the finished product is a multistep process comprising preparation of intermediate, pre-blending, granulation, post granulation blending and lubrication, film-coating and packaging. |
| Tablet / Capsule Image |

|
| Appearance | Slightly yellowish to greyish green, oblong-shaped tablets debossed with “AR 60” on one side |
| Imprint code / Engraving / Debossment | Debossed with “AR 60” on one side |
| Score | No Score |
| Color | Slightly yellowish to greyish green |
| Shape | Oblong-shaped |
| Dimension | 16.7 mm long x 8.7 mm wide |
| Mfg by |
Janssen Ortho LLC Gurabo, PR 00778 |
| Mfg for |
Janssen Products, LP Horsham, PA 19044 |
| Marketed by | - |
| Distributed by | - |
| Application No. | Prod No | Patent No | Patent Expiration | Drug Substance Claim | Drug Product Claim | Patent Use Code | Delist Requested | Link |
|---|---|---|---|---|---|---|---|---|
| N210951 | 1 | 10052314 | September 23, 2033 | - | - | U-2381 | - | Download |
| N210951 | 1 | 8445507 | September 15, 2030 | DS | DP | U-2237 | - | Download |
| N210951 | 1 | 8802689 | March 27, 2027 | - | - | U-2237 | - | Download |
| N210951 | 1 | 9388159 | March 27, 2027 | DS | DP | - | - | Download |
| N210951 | 1 | 9481663 | June 4, 2033 | DS | DP | U-2237 | - | Download |
| N210951 | 1 | 9884054 | September 23, 2033 | - | - | U-2237 | - | Download |
| N210951 | 1 | 9987261 | March 27, 2027 | - | DP | - | - | Download |
| USP Apparatus | Speed (RPMs) | Medium | Volume (mL) | Recommended Sampling Times (minutes) | Date Updated |
|---|---|---|---|---|---|
| USP Apparatus II | 75 RPM | 0.25% (w/v) SLS in 0.05 M Sodium Phosphate Buffer, pH 4.5 | 900 mL | Q in 30 min | As per SBOA |
| Label | Link |
|---|---|
| FDA label | Download |
| FDA chemistry review | Download |
| FDA Pharmacology Review(s) | Download |
| FDA Clinical Pharmacology Biopharmaceutics Review(s) | Download |
| FDA BE Recommendation | Download |
| European Public Assessment Report | Download |
| Territory | Brand name / Generic company name | Link |
|---|---|---|
| EU | ERLEADA | Download |
| UK | ERLEADA | Download |
| US | ERLEADA | Download |
| Date of first authorisation/renewal of the authorisation in EU: 14 January 2019 |
| www.accessdata.fda.gov, www.drugbank.ca, www.ema.europa.eu, www.medicines.org.uk, dailymed.nlm.nih.gov |